
GEO轉錄組芯片數據分析全流程:無gene symbol的R處理技巧(以GSE69063為例)
GEO數據庫轉錄組芯片數據處理與R分析:以GSE65682為例中我們提到過,GEO數據集如果只包含探針ID和對應的ENTREZID,則需要先將探針ID 轉換為ENTREZID,再將ENTREZID轉換為gene symbol。那么具體是需要進行什么操作呢?以數據集GSE69063為例,為大家詳細演示下如何使用R script窗口的腳本進行下載和分析。
1.數據集獲取
我們先進入GEO網站官網,根據GEO數據庫轉錄組芯片數據處理與R分析:以GSE65682為例中的方法找到數據集GSE69063的注釋文件GPL19983。

點擊GPL19983后,將頁面滾動至“Data table header descriptions”部分,發現注釋文件中只有探針ID和ENTREZID,并沒有我們需要的gene symbol,那這個時候我們該怎么處理呢?其實處理方法很簡單,只需要先將探針ID 根據注釋文件轉換為ENTREZID,再將ENTREZID轉換為gene symbol。
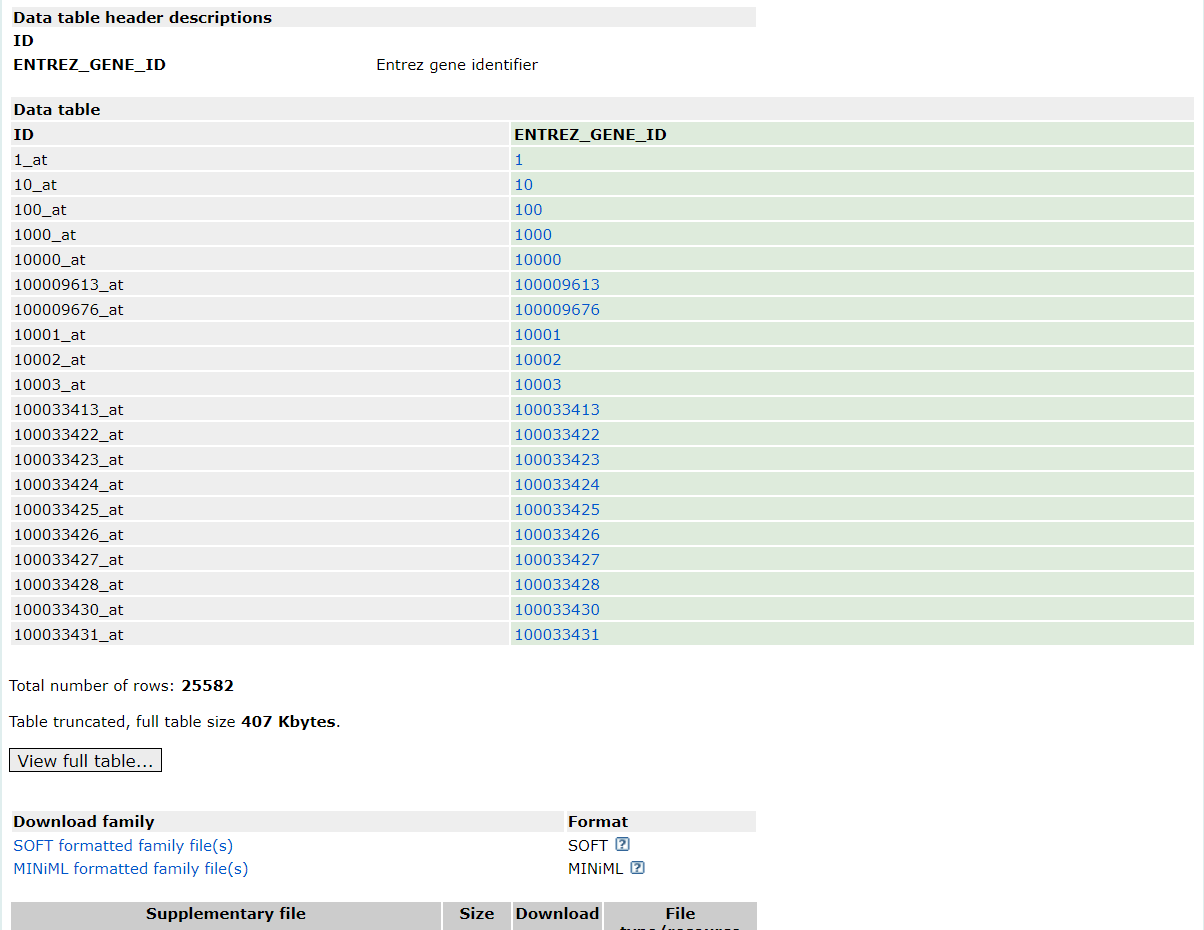
2. 數據下載與處理
獲取以上信息后即可直接進入R
獲取數據
#下載安裝相關的R包; #library(BiocManager) #install("GEOquery") #加載所需R包; library(GEOquery) library(limma) library(affy) library(data.table) library(dplyr) # 從GEO數據庫下載GSE69063數據集 # destdir="." 表示下載到當前工作目錄 # AnnotGPL = T 表示下載注釋文件 # getGPL = T 表示獲取平臺信息 # GSEMatrix = T 表示以GSEMatrix格式獲取數據 gset <- getGEO('GSE69063', destdir=".", AnnotGPL = T, ## 注釋文件 getGPL = T, GSEMatrix = T) ## 平臺文件 # 獲取表達矩陣(基因表達數據) exp <- exprs(gset[[1]]) # 獲取樣本的臨床信息 cli <- pData(gset[[1]]) ## 獲取臨床信息 # 獲取平臺注釋信息(探針與基因的對應關系) GPL <- fData(gset[[1]]) ## 獲取平臺信息
獲取到的表達矩陣行名為芯片的ID,列名為樣本ID。
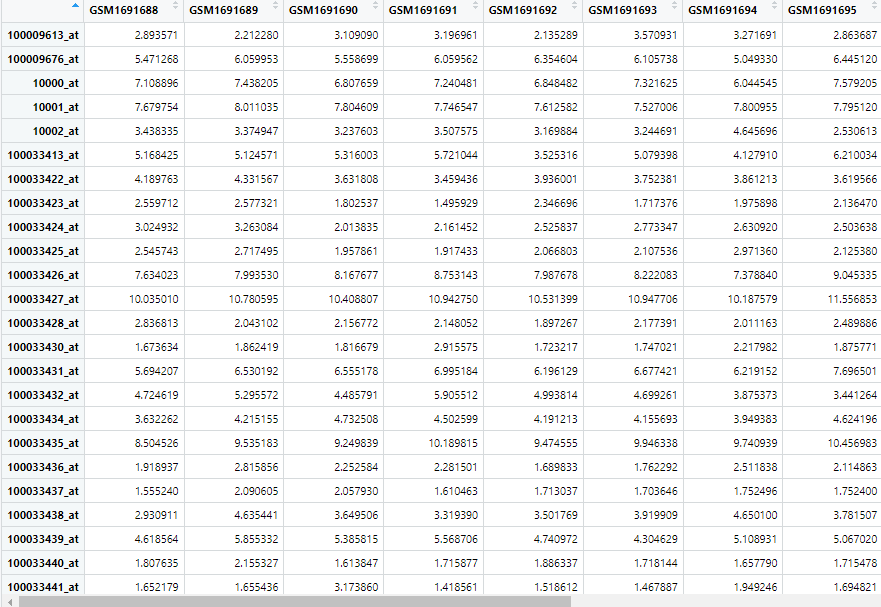
獲取到的臨床信息和注釋文件
臨床信息

注釋文件
探針ID到ENTREZ ID的轉換
# 從平臺信息中提取探針ID和ENTREZ基因ID列 gpl <- GPL[, c("ID", "ENTREZ_GENE_ID")] # 創建新的ENTREZID列,從ENTREZ_GENE_ID列提取數據 gpl$"ENTREZID" <- data.frame(gpl$"ENTREZ_GENE_ID", stringsAsFactors = F)[, 1] # 將表達矩陣轉換為數據框格式 exp <- as.data.frame(exp) # 在表達矩陣中添加探針ID列,使用行名作為探針ID exp$ID <- rownames(exp) # 將表達矩陣與平臺注釋信息按探針ID合并,添加ENTREZ基因ID信息 exp_ENTREZ <- merge(exp, gpl, by = "ID") # 移除包含NA值的行 exp_ENTREZ <- na.omit(exp_ENTREZ)
ENTREZ ID到基因符號的轉換
# 加載clusterProfiler包,用于基因ID轉換 library(clusterProfiler) # 使用bitr函數將ENTREZ基因ID轉換為基因符號(SYMBOL) # fromType = "ENTREZID" 指定輸入ID類型為ENTREZ # toType = "SYMBOL" 指定輸出ID類型為基因符號 # OrgDb = "org.Hs.eg.db" 指定使用人類基因注釋數據庫 gene_symbol <- bitr(exp_ENTREZ$ENTREZID, # 需要轉換的基因ID fromType = "ENTREZID", # 需要轉換的類型 toType = c("SYMBOL"), # 需要轉換為的類型 OrgDb = "org.Hs.eg.db") # 注釋包 # 將表達數據與基因符號信息按ENTREZID合并 exp_symbol <- merge(exp_ENTREZ, gene_symbol, by = "ENTREZID")
數據整理和去重處理
# 將數據框轉換為data.table格式,便于數據處理 df <- exp_symbol setDT(df) # 移除ENTREZ_GENE_ID列,因為已經有ENTREZID列 df1 <- df[, !(names(df) == "ENTREZ_GENE_ID"), with = FALSE] # 將data.table轉換回數據框格式 df1 <- as.data.frame(df1) # 將基因符號設置為數據框的行名 rownames(df1) <- df1$SYMBOL # 檢查基因符號的重復情況 table(duplicated(df1$"SYMBOL"))
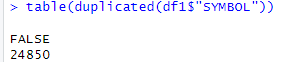
# 對重復的基因符號取平均值(去重處理) # 移除SYMBOL列(最后一列),然后對重復的基因符號取平均值 exp_unique1 <- avereps(df1[, -c(ncol(df1))], ID = df1$"SYMBOL") # 將處理后的唯一基因表達矩陣保存為CSV文件 write.csv(exp_unique1, "GSE69063_exp_unique.csv")
最后得到的exp_unique1行名為gene symbol,第一列為ENTREZID,第二列為探針ID,后面的數據就是這個基因在每個樣本中的表達水平了。

感謝大家的觀看!如果你在學習過程中遇到任何疑問,或者想要進一步探討相關話題,歡迎隨時私信我,或者在評論區留言交流。如果你覺得這個教程對你有所幫助,希望你能動動手指點個贊,收藏起來。

 浙公網安備 33010602011771號
浙公網安備 33010602011771號