
Nature Genetics | 本周最新文獻速遞
文章標題:A pangenome of maize provides genetic insights into drought resistance
中文標題:破譯玉米抗旱密碼!構建超級泛基因組揭示關鍵基因
關鍵詞:玉米、泛基因組、干旱脅迫、結構變異、基因功能
摘要總結:干旱是全球農業生產面臨的嚴峻挑戰,嚴重威脅著玉米等主要作物的產量和穩定性。深入解析作物抗旱的遺傳基礎,對于培育耐旱新品種、保障糧食安全至關重要。這篇文章通過對25個在抗旱性方面表現出顯著差異的玉米種質資源進行高質量的全基因組從頭組裝,并結合已有的31個玉米基因組序列,構建了一個包含56個種質的、迄今為止最全面的玉米泛基因組圖譜。基于這一強大的基因組資源,研究人員系統地進行了泛基因組分析、結構變異(SV)鑒定和全基因組關聯研究(GWAS),探索了玉米中廣泛的遺傳變異如何影響其在不同生長階段(苗期和開花期)的抗旱能力,并深入挖掘了控制這一復雜性狀的關鍵基因和分子機制。研究發現,在脫落酸(ABA)相關或干旱相關基因中存在著豐富的稀有等位基因變異和廣泛的調控多樣性,這些可能是導致不同種質間抗旱能力差異的重要原因。更重要的是,文章成功鑒定并功能驗證了三個新的抗旱關鍵基因:ZmUGE2、ZmSIL2_和_ZmASI3。功能實驗表明,_ZmUGE2_通過加強細胞壁的機械支撐來增強抗旱性;_ZmSIL2_作為一個轉錄因子,調控脅迫響應基因的表達;而_ZmASI3_則在干旱條件下協調雌雄穗的發育,減少“花期不遇”現象,從而降低產量損失。這對于深入理解植物抗旱的遺傳調控網絡、為玉米的分子設計育種提供寶貴的基因資源和理論依據,以及最終加速培育抗旱玉米新品種以應對全球氣候變化具有重要意義。

文章的亮點:
- 資源構建的全面性:構建了當時最全面的玉米泛基因組(包含56個高質量基因組),極大地擴展了玉米的遺傳變異圖譜,為深入研究玉米復雜性狀提供了前所未有的資源。
- 多階段抗旱機制解析:研究不僅關注了苗期的抗旱性,還重點解析了對產量影響更為關鍵的開花期抗旱機制,提供了更完整的抗旱調控網絡視圖。
- 關鍵新基因的發現與驗證:成功鑒定并系統地驗證了三個全新的、在不同層面發揮作用的抗旱基因(ZmUGE2、ZmSIL2、ZmASI3),揭示了細胞壁力學、脅迫信號轉導和生殖發育協同調控的新機制。
- 育種應用價值巨大:發現的稀有有利等位變異和關鍵基因為玉米抗旱育種提供了精確的分子標記和改良靶點,具有很強的實際應用潛力。
文章的局限:
- 群體水平SV基因分型的局限性:在較大的關聯群體中對泛基因組的所有結構變異進行精確分型仍然具有挑戰性,當前研究主要依賴于短讀長測序比對到線性參考基因組,這可能導致對復雜結構變異的檢測不準確或遺漏。
- 功能驗證的范圍有限:盡管文章對三個關鍵基因進行了深入的功能驗證,但泛基因組分析揭示的大量稀有等位變異和調控多樣性的具體生物學功能仍有待進一步的實驗驗證。
- 環境互作的復雜性:研究主要在控制條件下進行,而田間自然干旱環境更為復雜多變。基因型與環境的互作效應可能影響這些基因在不同生態條件下的實際表現,需要更廣泛的田間試驗。
文章標題:Genetic diversity and evolution of rice centromeres
中文標題:揭秘水稻著絲粒的遺傳多樣性與進化驅動力!
關鍵詞:水稻、著絲粒、遺傳多樣性、進化、逆轉錄轉座子
摘要總結:著絲粒是確保染色體在細胞分裂過程中正確分離的關鍵結構,其動態變化是真核生物進化和物種形成的核心驅動力,但其序列的高度重復性給研究帶來了巨大挑戰。這篇文章通過對來自Oryza AA基因組群的67個水稻基因組進行組裝,并分析了超過800個近乎完整的著絲粒序列,探索了水稻著絲粒的遺傳多樣性、結構層次和進化機制。研究人員開發了一套新的分析框架,通過從頭注釋著絲粒衛星序列CEN155,并采用漸進式壓縮策略,精確量化了水稻衛星陣列的局部同源化和多層嵌套結構。研究結果表明,水稻著絲粒的遺傳創新主要來源于結構變異和“嗜著絲粒”逆轉錄轉座子的插入。與染色體臂區相比,著絲粒區的單堿基替換率相對較低。通過對著絲粒CEN155陣列、逆轉錄轉座子和功能性著絲粒(由CENH3蛋白結合區域定義)的比較分析,揭示了它們之間動態但又相互關聯的演化關系。與擬南芥著絲粒進化的KARMA模型不同,該研究提出了一個新的假說:逆轉錄轉座子的入侵可能促進了祖先著絲粒衛星陣列的衰退,并推動了著絲粒的重定位。這一假說得到了CENH3染色質免疫共沉淀測序信號在天然衛星陣列之外區域富集的證據支持。這對于深入理解植物著絲粒的結構復雜性、動態演化規律及其在物種形成中的作用具有重要意義,為研究染色體生物學和進化遺傳學提供了新的視角和寶貴的數據資源。
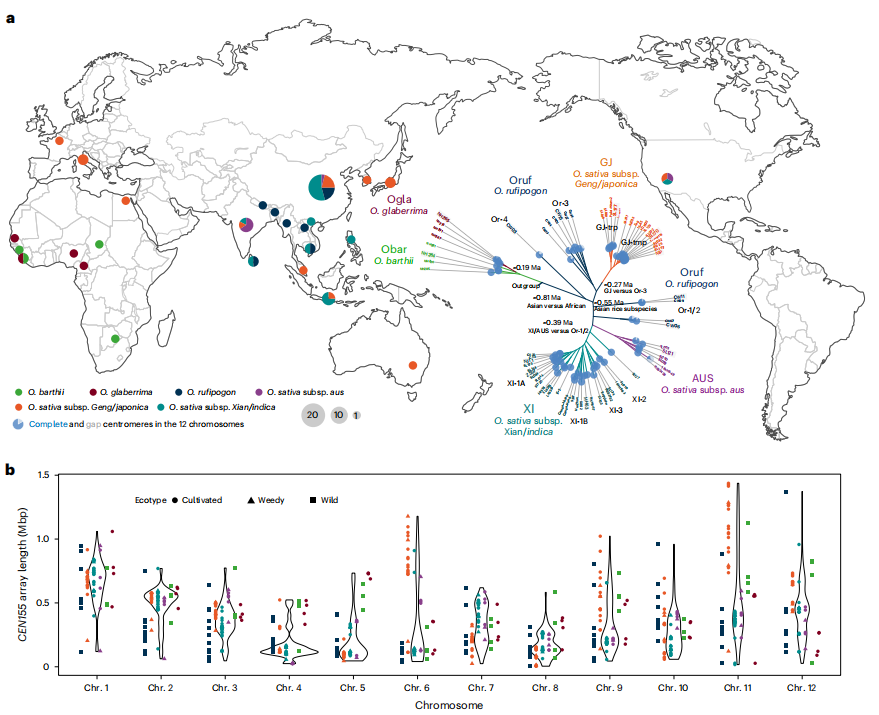
文章的亮點:
- 前所未有的規模和完整性:首次對Oryza AA基因組群的70個高質量基因組進行了近乎完整的著絲粒組裝和分析,揭示了前所未有的著絲粒遺傳多樣性。
- 創新的分析方法:開發并應用了一套新的分析框架,包括漸進式壓縮策略,能夠有效解析高度重復的著絲粒序列的局部同源化和多層嵌套結構。
- 提出了新的進化模型:基于詳實的數據,對經典的著絲粒進化模型提出了挑戰,提出了一個由逆轉錄轉座子入侵驅動的“祖先衛星陣列衰退-著絲粒重定位”新假說。
- 遺傳與表觀遺傳的關聯:結合了基因組序列分析和CENH3 ChIP-seq數據,關聯了著絲粒的序列結構與其功能(動粒附著位點),揭示了遺傳與表觀遺傳在著絲粒進化中的協同作用。
文章的局限:
- 物種范圍的局限性:研究主要集中在Oryza AA基因組群,其結論是否能推廣到更廣泛的植物界,特別是那些具有不同類型著絲粒(如全著絲粒)的物種,尚需進一步研究。
- 進化時間尺度的推斷:雖然研究揭示了動態的演化模式,但對于這些變化的精確時間尺度和選擇壓力等進化參數的推斷仍較為初步。
- 功能驗證的缺乏:研究提出的關于逆轉錄轉座子功能的假說主要基于相關性分析和計算推斷,缺乏直接的實驗證據來驗證其在著絲粒衰退和重定位中的因果作用。
文章標題:Complete genome assemblies of two mouse subspecies reveal structural diversity of telomeres and centromeres
中文標題:首次實現小鼠端粒到端粒的完整基因組組裝
關鍵詞:小鼠基因組、端粒到端粒(T2T)、結構多樣性、著絲粒、端粒
摘要總結:小鼠是理解哺乳動物疾病生物學的關鍵模型,但其參考基因組發布二十多年來仍存在大量缺口,尤其是在端粒和著絲粒等高度重復區域,限制了對這些關鍵結構域的研究。這篇文章通過采用單分子超長讀長測序技術,探索了兩個關鍵近交系小鼠亞種(C57BL/6J和CAST/EiJ)基因組的完整結構,并成功構建了首個小鼠端粒到端粒(T2T)的完整基因組。這些T2T基因組不僅填補了現有參考基因組(GRCm39)中的所有常染色體缺口,還首次完整地呈現了所有常染色體的端粒和著絲粒序列,為參考基因組新增了超過213Mb的新序列和517個新的蛋白質編碼基因。研究發現,這兩個亞種在端粒和著絲粒的大小及結構組織上表現出顯著的變異性。此外,文章還對兩個重要但之前不完整的基因座——性染色體上的假常染色體區(PAR)和KRAB鋅指蛋白基因座——進行了深入分析。研究揭示了PAR邊界在不同品系間的差異、節段性重復的拷貝數和大小變化,以及PAR基因中的大量氨基酸替換突變。這對于全面理解哺乳動物基因組的結構、功能和進化,特別是染色體末端和中心區域的生物學,以及解析與這些區域相關的疾病遺傳基礎具有重要意義,為未來的功能實驗和進化分析提供了前所未有的高質量基因組資源。

文章的亮點:
- 里程碑式的技術突破:首次實現了小鼠這一重要模式生物的端粒到端粒(T2T)完整基因組組裝,解決了長期以來困擾基因組學領域的高度重復區域組裝難題。
- 揭示了巨大的結構多樣性:通過比較兩個不同亞種的T2T基因組,揭示了在著絲粒、端粒、假常染色體區(PAR)等關鍵功能區域存在著驚人的結構變異和序列多樣性。
- 填補了基因組的“暗物質”:為小鼠參考基因組增加了大量新序列和數百個新基因,極大地豐富了我們對小鼠基因組內容和功能的認識,為研究這些新發現基因的功能鋪平了道路。
- 提供了寶貴的比較基因組學資源:高質量的T2T基因組為研究染色體進化、物種形成、基因組不穩定性以及與重復序列相關的疾病提供了精確的模型和工具。
文章的局限:
- Y染色體的缺失:盡管常染色體和X染色體達到了T2T級別,但Y染色體由于其更加復雜的重復結構,在此次研究中仍未能完全組裝,是基因組中尚待完成的部分。
- 品系的局限性:研究僅限于兩個特定的近交系(C57BL/6J和CAST/EiJ),雖然它們代表了不同的亞種,但仍無法完全覆蓋小鼠物種內廣泛的遺傳多樣性。
- 功能注釋的初步性:新發現的517個蛋白質編碼基因的功能注釋主要基于序列同源性預測,其真實的生物學功能和在生理病理過程中的作用需要大量的后續實驗驗證。
文章標題:Genotyping sequence-resolved copy number variation using pangenomes reveals paralog-specific global diversity and expression divergence of duplicated genes
中文標題:基于泛基因組解析拷貝數變異的基因分型揭示旁系同源基因特異性的全球多樣性及重復基因的表達差異
關鍵詞:拷貝數變異、泛基因組、基因分型、ctyper、旁系同源基因
摘要總結:拷貝數變異(CNV)基因在進化和疾病中扮演著重要角色,但其內部的序列變異在傳統大規模研究中仍是一個盲點。這篇文章利用泛基因組資源,開發了一種名為ctyper的新方法,探索了如何從二代測序樣本中精確解析CNV基因的等位基因特異性拷貝數和局部單倍型。ctyper方法能夠有效地處理包括非參考重復、基因轉換和復雜重排在內的復雜變異。在對3,351個CNV基因和212個醫學相關的挑戰性基因(CMR基因)的基準測試中,ctyper在CNV基因中實現了對96.5%的定相變異的捕獲,拷貝數正確率超過99.1%;在CMR基因中,這一比例也達到了94.8%。該方法計算效率高,在單個CPU上僅需1.5小時即可完成一個基因組的分型。應用ctyper的分型結果,研究發現其對基因表達的預測能力相比已知的eQTL變異提升了4.81倍。進一步分析揭示,7.94%的旁系同源基因存在顯著的表達分歧,4.68%存在組織特異性的表達偏好。例如,研究發現_SMN1_基因轉換為_SMN2_導致_SMN2_表達量降低,這可能影響脊髓性肌萎縮癥的病理;同時,_AMY2B_基因的易位重復導致其表達量增加。這對于在生物銀行(biobank)規模上實現對CNV和CMR基因的精確分型具有重要意義,為深入理解重復基因的全局多樣性、表達分歧及其在復雜疾病中的作用提供了強有力的工具。

文章的亮點:
- 方法學創新:開發了名為ctyper的新型基因分型工具,首次實現了利用泛基因組對復雜CNV區域進行序列解析級別的等位基因特異性拷貝數分型。
- 高精度與高效率:在包括醫學相關基因在內的大量復雜基因上展示了極高的準確性(>99%)和檢出率(>94%),同時保持了適用于大規模生物銀行項目的高計算效率。
- 揭示了新的生物學見解:通過對等位基因特異性表達的量化,揭示了旁系同源基因間廣泛存在的表達分歧和組織特異性偏好,并以_SMN1/SMN2_和_AMY2B_為例,展示了其在疾病相關基因中的潛在功能影響。
- 提升了基因表達預測能力:證明了序列解析的CNV分型結果能顯著提高對基因表達水平的預測能力,遠超傳統eQTL變異的效果,強調了精細遺傳變異在調控網絡中的重要性。
文章的局限:
- 依賴于泛基因組的完整性:ctyper的性能高度依賴于所使用的泛基因組參考數據庫的質量和全面性。如果某個樣本中存在數據庫未包含的稀有或新的單倍型,分型準確性可能會下降。
- 亞群粒度的限制:由于樣本量的限制,當前的關聯分析主要基于較大的“亞群”(subgroups)而非單個等位基因(PAs),這可能掩蓋了更精細等位基因之間的功能差異。
- 短讀長測序的固有局限:盡管該方法針對二代測序數據進行了優化,但在極度復雜或超長重復區域,短讀長數據本身的信息含量仍然有限,可能導致分型模糊或錯誤。
- 表達分析的局限:eQTL分析僅限于能夠被RNA-seq明確映射的基因,對于那些轉錄本高度相似以至于無法區分的旁系同源基因,其表達量只能合并分析,限制了對個體旁系基因表達調控的解析。
文章標題:Locityper enables targeted genotyping of complex polymorphic genes
中文標題:Locityper實現對復雜多態性基因的精準靶向分型
關鍵詞:基因分型、多態性基因、泛基因組、Locityper、結構變異
摘要總結:人類基因組中包含數百個與疾病相關的、結構高度可變的多態性基因座,這些區域因其復雜性而難以通過標準方法進行準確的變異檢測。這篇文章通過開發一種名為Locityper的新工具,探索了如何利用短讀長和長讀長全基因組測序數據,對這些具有挑戰性的基因進行靶向分型。Locityper首先從泛基因組等資源中提取目標基因座的已知單倍型,然后招募并比對測序讀長至這些單倍型上,通過優化讀長比對、插入片段大小和讀長深度分布等多個維度的信息,找到最可能的單倍型組合。在對256個醫學相關的挑戰性基因座的測試中,無論是使用短讀長還是長讀長數據,Locityper的分型質量值(QV)中位數均超過35,其性能分別比當前頂尖的Illumina和PacBio HiFi變異檢測流程高出10.9和1.7個點。此外,Locityper還能有效處理如HLA、KIR、MUC和FCGR等超多態性基因家族。由于其高效的計算性能,Locityper能夠擴展到生物銀行規模的隊列研究中。這對于開啟對以往難以分析的疾病相關基因的關聯研究具有重要意義,為揭示復雜疾病的遺傳基礎提供了強大的新工具。

文章的亮點:
- 技術創新性:提出了一種新的靶向基因分型策略,整合了讀長比對、插入片段大小和深度等多源信號,專門用于解析傳統方法難以處理的高度多態和結構復雜的基因座。
- 高性能與廣適用性:在短讀長和長讀長數據上均表現出超越現有主流變異檢測流程的卓越準確性,證明了其在不同測序平臺上的強大適用性。
- 攻克關鍵醫學基因:成功應用于HLA、KIR、MUC等多個以極度多態性著稱的、與免疫和多種疾病密切相關的基因家族,展示了其在醫學研究中的巨大潛力。
- 可擴展性強:算法計算效率高,使其能夠輕松擴展到數萬甚至數十萬樣本的生物銀行級別隊列,為這些大規模數據集中“不可見”的遺傳變異研究鋪平了道路。
文章的局限:
- 依賴于參考單倍型庫:Locityper的準確性依賴于一個全面的單倍型參考面板。如果一個樣本攜帶的單倍型在參考庫中不存在或代表性不足,分型準確性會顯著下降。
- 無法發現新單倍型:該工具的設計目標是“識別”已知的單倍型,而不是“發現”新的單倍型。它只能將樣本的序列匹配到參考庫中最相似的單倍型,而不能從頭構建新的等位基因。
- 對旁系同源基因的區分能力有限:當兩個或多個高度同源的基因座(旁系同源基因)在基因組中物理位置接近時,可能會存在讀長招募的交叉干擾,影響分型的準確性。
- 在極長或高度重復區域的挑戰:盡管性能優越,但在處理含有極長串聯重復(如某些MUC基因的VNTR區域)的基因座時,短讀長數據仍然存在固有的局限性,可能導致分型精度下降。
文章標題:Real-time dynamic polygenic prediction for streaming data
中文標題:實時動態多基因風險預測模型(rtPRS-CS)問世
關鍵詞:多基因風險評分、精準醫療、流式數據、動態預測、rtPRS-CS
摘要總結:多基因風險評分(PRS)是推動精準醫療的重要工具,但現有方法依賴于靜態的全基因組關聯研究(GWAS)摘要統計數據,更新周期長,無法充分利用醫療保健領域持續產生的新基因和健康數據。這篇文章通過在PRS-CS框架基礎上進行擴展,開發了一種名為實時PRS-CS(rtPRS-CS)的新方法,探索了如何隨著每個新樣本的收集,在線、動態地優化和標準化PRS,從而實現對后續患者風險的更精準預測。rtPRS-CS利用隨機梯度下降算法,在每個新樣本加入時迭代更新SNP權重,整個過程計算高效。研究通過廣泛的模擬評估了rtPRS-CS在不同遺傳結構和訓練樣本量下的性能。利用兩個大規模生物銀行(MGBB和UKBB)的定量性狀數據,研究表明rtPRS-CS能夠整合海量流式數據,隨時間推移顯著提升PRS的預測能力。此外,研究還將rtPRS-CS應用于包含七個亞洲地區的22個精神分裂癥隊列,證明了該方法在動態捕捉不同遺傳背景人群的健康狀況變化和預測疾病風險方面的臨床效用。這對于在真實世界的醫療環境中,最大化利用持續增長的數據以提供最準確的遺傳風險預測具有重要意義,推動了PRS從靜態研究工具向動態臨床決策支持工具的轉變。

文章的亮點:
- 方法學上的范式轉變:首次提出并實現了一個實時、動態更新的多基因風險評分(PRS)框架,打破了傳統PRS依賴靜態、周期性更新的GWAS摘要統計的局限。
- 高效的在線學習算法:巧妙地運用隨機梯度下降算法,使得模型可以在每個新樣本加入時進行快速、輕量級的權重更新,極大地提高了計算效率和應用的時效性。
- 跨人群和跨性狀的穩健性:通過在模擬數據、兩個大型生物銀行的多種定量性狀以及跨越多個亞洲人群的精神分裂癥隊列中的成功應用,證明了該方法的強大穩健性和廣泛適用性。
- 動態適應真實世界場景:模型不僅能整合新樣本以提高預測準確性,還能動態調整和標準化PRS,以適應人群結構變化和個體健康狀態的動態變化(如從健康對照轉為病例),更貼近復雜的臨床現實。
文章的局限:
- 對初始模型參數的依賴:rtPRS-CS的性能在一定程度上依賴于初始基線GWAS提供的全局和局部遺傳度參數。如果初始參數估計不準(尤其是在小樣本基線研究中),可能會影響后續更新的效率和最終的準確性。
- 樣本順序和相關性的潛在影響:盡管研究表明最終結果與樣本順序無關,但在訓練過程中,樣本的加入順序以及樣本間的親緣關系可能導致預測評分的短期波動,這在臨床應用中需要謹慎處理。
- 對復雜遺傳結構的擴展性:當前模型主要適用于相對同源的歐洲人群,雖然在亞洲人群中也表現良好,但對于高度混合或跨大陸人群的直接應用仍具挑戰,需要與PRS-CSx等跨人群方法進一步整合。
- 無法替代周期性GWAS:該方法是一種高效的“增量更新”工具,但不能完全替代對表型和基因型數據進行嚴格質控和協調的周期性大規模GWAS,后者對于校正批次效應和獲得最精確的效應量估計仍然至關重要。
文章標題:An African ancestry-specific nonsense variant in CD36 is associated with a higher risk of dilated cardiomyopathy
中文標題:非洲裔人群擴張性心肌病高風險之謎揭曉:CD36基因變異是關鍵
關鍵詞:擴張性心肌病、非洲裔、CD36、全基因組關聯研究、功能喪失變異
摘要總結:擴張性心肌病(DCM)在非洲裔人群中負擔尤為嚴重,其背后的遺傳原因尚不完全清楚。這篇文章通過對包含1,802名非洲裔DCM病例和93,804名對照的大規模全基因組關聯研究(GWAS),探索了導致非洲裔人群DCM風險升高的特異性遺傳因素。研究發現,_CD36_基因中的一個無義變異(rs3211938:G )與DCM風險顯著增加相關。該變異在非洲裔人群中頻率較高(約17%),但在歐洲裔人群中幾乎不存在(<0.1%),這可能與它在瘧疾抵抗中提供的保護性選擇優勢有關。攜帶該風險等位基因純合子的個體(約占非洲裔人群的1%),其DCM風險增加了約三倍。在沒有臨床心肌病的個體中,純合子也表現出左心室射血分數的顯著降低,呈現亞臨床表型。該單一變異對非洲裔人群DCM的群體歸因分數達到8.1%,解釋了與歐洲裔相比約20%的額外DCM風險。進一步的實驗驗證,利用人誘導多能干細胞來源的心肌細胞模型證明,_CD36_基因的功能喪失會損害心肌細胞對脂肪酸的攝取,破壞心臟的能量代謝和收縮功能。這對于揭示非洲裔人群中DCM的一個重要且普遍的遺傳病因,闡明心肌能量代謝受損作為DCM發病機制的核心環節,以及為針對特定人群的DCM風險篩查和潛在治療干預具有重要意義。

文章的亮點:
- 重大發現:首次發現了一個在非洲裔人群中常見、但在其他人群中罕見的_CD36_無義變異是DCM的主要風險因素,為解釋該人群中的健康不平等現象提供了重要的遺傳學證據。
- 清晰的機制闡釋:通過“GWAS-精細定位-功能實驗”的完整研究鏈,清晰地證明了_CD36_功能喪失導致心肌脂肪酸代謝障礙,進而引發收縮功能障礙,最終導致DCM的病理生理通路。
- 進化與疾病的關聯:將該風險變異與瘧疾抵抗的陽性選擇壓力聯系起來,為“進化權衡”(evolutionary trade-off)理論提供了一個生動的實例,即一個在特定環境下有利的基因變異可能在另一環境下導致疾病風險。
- 臨床轉化潛力:研究結果提示_CD36_變異可作為非洲裔人群DCM風險分層的重要生物標志物,并且為開發靶向心肌能量代謝的治療策略提供了新的思路。
文章的局限:
- 病例定義的局限性:DCM病例的確定主要基于電子健康記錄中的診斷代碼,可能存在診斷不精確或分類錯誤的情況,但這通常會使關聯結果偏向于無效假設,從而低估了真實的關聯強度。
- 未考慮其他遺傳因素:在評估該變異對人群風險差異的貢獻時,研究未全面考慮其他可能存在的、在不同人群中頻率各異的DCM風險或保護性遺傳變異。
- 功能實驗模型的簡化:體外hiPSC心肌細胞模型雖然揭示了核心分子機制,但它使用的是siRNA介導的完全敲低,而非直接引入rs3211938變異,且無法完全模擬體內復雜的生理環境和長期的代償與失代償過程。
- 成像隊列樣本量較小:用于驗證亞臨床表型的多中心影像學隊列中,攜帶純合風險變異的個體數量較少,盡管結果一致且統計學顯著,但更大的樣本量將能提供更穩健的效應量估計。
致謝橙子牛奶糖(陳文燕),請用參考模版:We thank the blogger (orange_milk_sugar, Wenyan Chen) for XXX
感謝小可愛們多年來的陪伴, 我與你們一起成長~

本文來自博客園,作者:橙子牛奶糖(陳文燕),轉載請注明原文鏈接:http://www.rzrgm.cn/chenwenyan/p/19191259

 浙公網安備 33010602011771號
浙公網安備 33010602011771號